






�����v���W�F�N�g
���{�k�N���I�v���e�I�~�N�X�����Ɍ�����LC-MS �V�X�e���̊J��
Development of a LC-MS system for ribonucleoproteome analysis
�c������1,4�@���R�m2,4�@�����M�O3,4�@�E�ӏr��1,4
Masato Taoka, Hiroshi Nakayama, Nobuhiro Takahashi, Toshiaki Isobe
1.��s��w���� ��w�@ ���H�w������ ���q�����Ȋw��U
2.�����w������ ������� ��[�Z�p��Օ��� �o�C�I��̓`�[��
3.�����_�H��w �����Ȋw�Z�p�����@ �����_�w����
4.CREST
�v�|
�ߔN�A�Q�m����̔�R�[�h�̈�Ȃǂ��琶����non-coding RNA�incRNA�j�ƃ^���p�N���̕����́iRNP�����́j���]�ʂ�|����n�߂Ƃ���זE�@�\�̒��߂ɏd�v�Ȗ������ʂ����Ă��邱�Ƃ����炩�ɂȂ��Ă���BRNP�����̂Ɋւ���]���̌����ł́A�^���p�N�������̓v���e�I�~�N�X�̕��@�ʼn�͂��ARNA�����̓^���p�N���Ƃ͓Ɨ��ɁA�t�]�ʔ����ŕϊ�����cDNA����͂���ԐړI�ȕ��@�ɂ���͂���Ă����B�M�҂�́A�ŐV�̎��ʕ��͖@�ɂ����RNP�����̂Ȃǂ��\������RNA���^���p�N���Ɠ����ɒ��ڃQ�m����ɋA�����ē��肵�A�]�ʌ�C�����܂މ��w�\���������x�ʼn�͂ł��郊�{�k�N���I�v���e�I�~�N�X�����̂��߂̊v�V�I�ȃv���b�g�t�H�[���̊J����i�߂Ă���B�{�e�ł́A���̕��@�̌����RNP�����̉�͂ւ̉��p�ɂ��ďЉ���B
�͂��߂�
�ߔN�̌����ɂ��ARNA�̓Q�m���ɕۑ����ꂽ��`���ƃ^���p�N�����q�����ԑ̂Ƃ��Ă̖����ɉ����āA���F�̂̈��萫����f�����O�A�]�ʂ�|��A�����A�����܂ޑ��ʂȍזE�@�\�̒��߂ɏd�v�Ȗ�����S���Ă��邱�Ƃ����炩�ɂ���Ă���B������RNA�̑����̓^���p�N�����R�[�h���Ȃ��Q�m����̔�R�[�h�̈�����̈�`�q�̃C���g��������]�ʂ���A���G�ȓ]�ʌ�C�������̂��A�����̃^���p�N�������ƕ����̂��`�����ċ@�\���邱�Ƃ��m���Ă���B�hnon-coding RNA�incRNA�j�h�Ƒ��̂���邱����RNA�́AmicroRNA��piwiRNA�Ȃǂ̃T�u�N���X�ɕ��ނ���邪�A������������̔����╪���Ȃǂ̍����@�\�̒��߂ɒ��ڊ֗^���邱�ƁA�܂����ُ̈�̓K����`�Ԍ`���s�S���e��̎����̌����ƂȂ邱�Ƃ�����Ă���B����������ncRNA�̌����́A�זE�@�\����͂����b�����w�����łȂ��A�K�����n�߂Ƃ��鎾�a�̑����f�f��ڎw���Տ�������A�n��̕W�I���邢�͈��i�̑f�ނƂ��Ă����ڂ���Ă���B
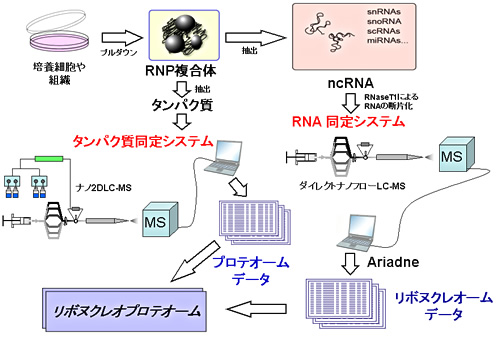
�Q�m������]�ʂ��ꂽncRNA�͂������ɓ���̃^���p�N���Q�ƕ����̂��`�����A���≖��̏C����v���Z�V���O�Ȃǂ̕��G�ȉߒ��ɂ���Đ��n���邱�Ƃ��m���Ă���B���������āAncRNA�̍\����@�\�A�������ߒ��𗝉����邽�߂ɂ́ARNA-�^���p�N�������́iRNP�����́j��P�ʂƂ��āA���̍זE���ł̃_�C�i�~�N�X���I�ɉ�͂��錤�����d�v�ł���B�M�҂�́A�v���e�I�~�N�X��RNA������Z�������@�\��RNP�����̂Ɋւ��錤�����u���{�k�N���I�v���e�I�~�N�X�v�ƌĂԂ��Ƃ��Ă��A���̂��߂̕��@�_�̊J����i�߂Ă���B�ŐV�̎��ʕ��͖@�𒆐S�Ƃ���RNA��͂̂��߂̊�{�I�ȃv���b�g�t�H�[���̊T�v���}�P�Ɏ������B���̕��@�ł́A�e�a���^�O�Ȃǂ𗘗p���čזE���番������RNP�����̂Ɋ܂܂��RNA���������{�k�N���A�[�[�ŏ������A��������RNA�f�Ђ̃^���f�����ʕ��̓f�[�^����Q�m�����𗘗p���ĖړI��RNA�肷��Ɠ����ɓ]�ʌ�C�����܂މ��w�\������͂��邱�Ƃ�ڕW�Ƃ��Ă���B�{�e�ł́ALC-MS�@�𗘗p�����]����RNA�����̊T�v�ƁA�M�҂炪�J����i�߂Ă���RNA��͂̂��߂̎��ʕ��̓v���b�g�t�H�[���̌���ɂ��ďЉ���B
�}1

LC-MS�@�𗘗p����RNA����
ncRNA���܂�RNA�̑����͓]�ʌ�ɓ���̓��≖��C������Đ��n���邱�Ƃ��m���Ă���B���݁A���j��������^�j�����ɂ����鐶�����RNA�ɂ��āA���Ȃ��Ƃ�107��ނ̏C��������Ă���(1)�B�����̏C���́A���f�t����`�����A�ِ����A����ւ̃A�~�m�_�ⓜ�̕t���A5�f���[�̃g�����`���O�A�j�����ȂǁA���l���ɕx��ł���B�]����RNA�C���̉�͂́A��Ƃ��čזE���ܗʂ̑���tRNA��rRNA�A���邢�͒ᕪ�q�j��RNA�isnRNA�j�ōs���Ă��邪�A������RNA�ł̏C���́A����q��e�\�͂̒��߂◧�̍\���̈��艻(2)�A�זE���Ǎ݂̒���(3)�A�R�������ϐ�(4)�ȂǁARNA���q�̍\���Ƌ@�\�̔����⒲�߂ɕK�{�ł��邱�Ƃ��m���Ă���B��ʂ�RNA�̓]�ʌ�C���́A��{�I�ɂS��ނ݂̂̍\���v�f����Ȃ�RNA�ɑ��l����t�^���A���̂����Ȃ��ޗ��ő��l�ȋ@�\�ƕ��G�Ȓ��߂��\�Ƃ��邽�߂̐헪�̂P�ɂȂ��Ă���ƍl������B�]�����猤�����i��ł���X�v���C�Z�I�\�[����{�\�[���Ȃǂ�RNP�����̂̐�������@�\�̒��߂ɂ́A�j����RNA�isnoRNA�j�Ȃǂɂ���ċK�肳��镔�ʓ��ٓI�ȃ��`������[�E���W�����Ȃǂ̋ɂ߂ĕ��G�Ő����ȓ]�ʌ�C�����ւ���Ă��邱�Ƃ������̌����ɂ���Ė��炩�ɂȂ��Ă���B���������āA�]�ʌ�C���̉�͂�RNA�̍\���Ƌ@�\�̌����ɂƂ��Ė{���I�ɏd�v�ȈӖ��������Ă���B
����A�]����RNA�����́A��Ƃ��ċt�]�ʔ����ɂ���č�������cDNA��PCR�@�ő������A����z�͑��u��A���C�@�ŕ��͂���Ԑږ@�ɂ���čs���Ă����B�����̕��@�͌����悭���ʂ�RNA����͂ł��邽�߁A�זE��RNA�������x�Ŗԗ��I�ɕ��͂���ړI�ɂ͓K���Ă��邪�A�t�]�ʍy�f�̓��ِ��ɗR�����邳�܂��܂Ȗ��_�ƂƂ��ɁA��ʓI�ȕ��͂�������Ƃ�]�ʌ�C���̉�͂��ł��Ȃ��Ȃǂ̑����̖��������Ă����BRNA�̎��ʕ��͖@�́A��Ƃ��āA���������]���@�̌��_��₤�]�ʌ�C���̉�͖@�̂P�Ƃ��ČÂ�����s���Ă���(5, 6)�B�ŋ߂̌����ł́A�Ⴆ�Γ�����w�̗��͎��ʕ��͖@�𗘗p���āA�@�\���m��`�q�Q����ԗ��I��RNA�C����`�q��T�����錤��(7)��ARNA�C���ُ�ɋN�����鎾���̌���(8)�ARNA�C�������肷��tRNA�̍זE���Ǎ݉��@�\�̌���(3)��ncRNA�Ɋ܂܂��RNA�C���̓���(9)�Ȃǂ̐��ʂ���Ă���B�܂��A�I���S�k�N���I�`�h�Ԃ̉���Ό`���\�𗘗p���ĕ�����RNA���q���ɒP����������Z�p���J����(10)�A���̕��@�Ǝ��ʕ��͖@��g�ݍ��킹�邱�ƂŁA�̎����זE�ɓ��ٓI�ɔ������ăR���X�e���[���⎉�b�_�̑�ӂɊ֗^���Ă���miRNA�imiR-122�j�̈��艻�Ɋ�^����A�f�j�����C��������ƂƂ��ɁA���̏C�����|��A�|�������[�[GLD-2�ɂ����̂ł��邱�Ƃ𖾂炩�ɂ��Ă���(11)�B
���{�k�N���I�v���e�I�~�N�X�����̂��߂�RNA���ʕ��͖@
RNA��LC-MS�@�̍����\��
�M�҂�́ARNP�����̂Ȃǂ��\������ᕪ�qRNA�ڕ��͂��邱�ƂŁA���ꂼ���RNA���Q�m�����ɋA�����A�]�ʌ�C�����܂߂��\���Ƌ@�\�̊W�𖾂炩�ɂ��邱�Ƃ�ڕW�Ƃ��Ă���B���̕��@�̑傫�ȓ����́A�����Ɋ܂܂��RNA�������Q�m����ɋA�����邱�ƂŁA�ړI�Ƃ���RNP�����̂Ɋ܂܂��RNA��\���m���ɂ���Ă��炩���ߗ\�����邱�ƂȂ��A���m�̕��q����܂߂ē���ł��邱�Ƃł���B���̕��@�̊J���ɂ������ĕM�҂�́A�v���e�I�~�N�X�����̂��߂ɊJ�������_�C���N�g�i�m�t���[LC-MS�V���b�g�K����̓V�X�e��(12)��RNA���͂̂��߂ɍœK�����邱�Ƃ���{�I�Ȑ헪�Ƃ������i�}�P�j�ARNA�̓^���p�N���ɔ�ׂč\�z�P�ʂ����Ȃ��ĉ��w�I�ȑ��l�������������ƁA�܂��^���p�N���̓Q�m���̖�Q���قǂ��߂�ORF�ɋK�肳���̂ɑ��ARNA�̓Q�m���̑S�̈悩�琶�����A�������]�ʗ̈悪���m�ɒ�`����Ă��Ȃ����ƂȂǂ���A���܂��܂Ȍ����⌤�����K�v�ł������B
RNA��LC�����Ɋւ��錤���ł́A�e��̋t���J�����ƕ��������̑g�ݍ��킹�ɂ��Č������A�����_��ɕx�މ������ł���RNA�ɑ��ĕێ��\�͂������V���J�nC-30�t���n�[�U�܂Ɖ����������RNA�ƃC�I���y�A���`������������̍��������}�̂̋����C�I����I�肷�邱�Ƃŕ������œK�����A�ᕪ�qRNA�₻��RNase�������������悭�����ł���i�mLC�@���J������(13)�B�܂��A���a150��m�̃G���N�g���X�v���[�J�����������i�mLC����50-100 nL/���̔������ő��t����Ă���RNA�n�o�t�ɃI�����C���ŘA���I�ɃA�Z�g�j�g������Y�����A���d�ׂ����e������RNA�������A�]���͍���Ƃ���Ă����u�l�K�e�B�u���[�h�v�ň��肵�ăC�I�������邽�߂̕⏕�X�v���[���u�����삵�đ��������B�����̌����ŊJ������LC�V�X�e�������ʐ��x���ɂ߂č����d��^�t�[���G�ϊ����ʕ��͌v�ƘA�����A����ɂ͓Ǝ��ɊJ�������z��f�[�^�x�[�X�����G���W���iAriadne�Ɩ����F��q�j�Ƒg�ݍ��킹�āARNA�������ǂ��������Ď��ʕ��́iMS�j����у^���f�����ʕ��́iMS/MS���́j���A�z��f�[�^�x�[�X�����\���I�ɓ����RNA��ł���v���g�^�C�vRNA��̓V�X�e�������삵���B
���̃V�X�e���́A���w��������siRNA�△�זE�����n�ō�������mRNA�Ȃǂ�RNase T1�ŏ������ē���ꂽRNA�f�Ёi�I���S���{�k�N���I�`�h�j�̐������ʕ��͂≖��g���̕��͂ɗL���ł��������ARNA�̕��͊��x�́A���l�̃V�X�e���ɂ���ă^���p�N���͂������̊��x�ɔ�ׂĖ�1/10���x�ł������B�����ŁA���̃V�X�e���������x�����邽�߁A RNA������RNase��������ESI�J�����ɓ������邽�߂Ɏg�p����u�g���b�v�J�����v��������A���������A����Ƀi�mLC����n�o����RNA�f�Ђ������悭�C�I�������邽�߂̕⏕�X�v���[���u�����ǂ����i�����o��ρj�B�܂��A�|�{�זE��RNP�����̂Ɋ܂܂��RNA��d�C�j���ŕ�����ɃQ������RNase��������LC-MS�V�X�e���ɓ������邽�߂̑O�����@���œK�������B�����̉��ǂɂ��A���݂̃V�X�e���ł͎���i�ɔ�ׂ�RNA�̕��͊��x����10�{���サ�A�|�{�זE�Ȃǂ���e�a���^�O�𗘗p���ĕ����������ʂ̃X�v���C�Z�I�\�[���i��q�j��{�\�[�����̂Ɋ܂܂��t�F���g�������x���̃^���p�N����RNA�������قړ����x�̊��x�œ��肵�A���̉���z���A���`�����Ȃǂ̏C�����܂߂����w�\�����^���p�N�������Ɠ����ɉ�͂��邱�Ƃ��ł���悤�ɂȂ���(13)�B����Ɍ��݂ł́A�����Ɋ܂܂��RNA�̕�������O�����ALC-MS���͂ƃf�[�^��͂Ɏ���V�X�e���̑S��������i�߂Ă���B
RNA�����̂��߂̃\�t�g�E�G�A
�O���ł��G�ꂽ�悤�ɁARNA���܂ފj�_�̉��w�\�������ʕ��͖@�ɂ���ĉ�͂��錤���͌Â�����s���Ă������ARNA�̎��ʕ��͏��Ƃ�킯�^���f�����ʕ��͖@�œ���ꂽRNA�f�Ђ�MS/MS���Q�m�����𗘗p���Ă��Ƃ�RNA����肵�A���̉��w�\������͂��鎎�݂͍s���Ă��Ȃ������B�M�҂�́A���l�̕��@���v���e�I�~�N�X�����ł͊��Ɉ�ʓI�ł���A���������̕��@��RNA�����ɓK�p�ł���A�Q�m�����𗘗p���ď]���͒m���Ă��Ȃ����m��RNA������ł��邱�ƁA�܂�RNA�̋@�\�����⒲�߂ɕs���ȓ]�ʌ�C����זE���ł̑�Ӊߒ��̉�͂��\�ɂȂ�ƍl�����B���̕��@���������邽�߂̒��S�ƂȂ�Z�p�̂P��RNA�̎��ʕ��͏����Q�m�����ƘA������\�t�g�E�F�A�A���Ȃ킿�Q�m�������G���W���ł������B����Ɠ��l�̋@�\�����\�t�g�E�G�A�́A�v���e�I�~�N�X�ł�Mascot��Sequest�ȂǂƂ��Ĉ�ʂɒm���Ă��邪�A�J�����͂��߂Ă݂�ƁA�^���p�N����ΏۂƂ����\�t�g�E�F�A�Ŏg�p����Ă���A���S���Y�������̂܂�RNA��͂ɓK�p���邱�Ƃ��ł��Ȃ����Ƃ��킩�����B���̎�ȗ��R�͎��̒ʂ�ł���B
1) RNA�ƃ^���p�N���͂������������|���}�[�ł��邪�A�^���p�N���͗����[�Ɠ���̑����݂̂ɉ𗣊�����i�A���M�j���A�q�X�`�W���A���W���̑����c��j�̂ɑ��āARNA�͎卽�ɉ𗣊�i�����_��j�������߁A�Փˉ𗣁iCID�j�@�ɂ��MS/MS���͂ł�RNA�̒f�Љ��v���t�@�C���̓y�v�`�h�Ƃ͑傫���قȂ�i��q�j�B
2)���݂̎��ʕ��͋Z�p�ł�RNA�Ȃǂ̍����q�����̂܂ܕ��͂��č\����͂��邱�Ƃ͍���ł��邽�߁ARNase�Ȃǂŏ������Đ�����RNA�f�Ђ̎��ʕ��͏����Ƃ�RNA����肵�A���̍\������͂��邱�Ƃ��K�v�ł���B�������Q�O��ނ̃A�~�m�_����\�������^���p�N���ɔ�ׂāARNA�͂S��ނ̃k�N���I�`�h�����݂̂���\������鑽�l���̏����������q�ł��邽�߁A���ʕ��͖@�ʼn�͉\�Ȕ�r�I������RNA�f�Ђ̔z���猳��RNA����肷�邱�Ƃ�����ł���B
3)�^���p�N���̓R�[�h�̈�̑啔����ORF�Ƃ��ċK�肳��Ă���A�z��DB���悭��������Ă���̂ɑ��āA��R�[�hRNA�ɑΉ�����Q�m��DNA�̓���͌������i�߂��Ă���i�K�ŁA����ł͗\��������ł���B���̂���RNA�肷�邽�߂ɂ͓Ǝ��̔�R�[�hRNA DB���\�z���邩�A�Q�m���z��S�̂�DB�Ƃ��Č������邱�Ƃ��K�v�ł���B
�����̖��_���������Ȃ���RNA�̌����G���W�����J������ړI�ŁA�M�҂�́A�܂��n�߂ɏՓˉ𗣖@�ɂ���ē�����RNA�f�Ђ̊J��p�^�[�����ڍׂɉ�͂����B����܂�RNA��MS/MS��͂͂قƂ�Ǎs���Ă��炸�ADNA�Ɠ��l���Ƃ����قȂ�Ƃ�������݂���ł���������(14, 15)�A����RNA��̂���P���������mRNA��MS/MS�X�y�N�g���f�[�^�����W���邱�ƂŁA�Փˉ𗣖@�ł�RNA�̒f�Љ����[���������I�ɉ�͂����B���̌��ʁA�J����卽�����_�����̂���c/y�ʂ�a/w�ʂ�2�ӏ����I��I�ɊJ�邱�Ƃ��킩�����i�}�Q�j�B�܂��ARNA�ł̓^���p�N���ƈقȂ�卽�ɉדd���ʂ����邽�߂ɓ����C�I�����������o����邱�ƁADNA�ł͉���̒E���������p�x�Ő�����̂ɑ���RNA�ł͂قƂ�lj���E���������Ȃ��ȂǁARNA�ɓ��قȊJ���l�������邱�Ƃ��킩�����B���̊J���l���̓G���N�g���X�v���[�C�I�����@��MALDI�@(16)�A���邢�̓C�I���g���b�v�^��n�C�u���b�h�^���ʕ��͑��u�Ȃǂ̕��@�Ɉˑ�������v���邱�Ƃ���A�Փˉ𗣖@�ɂ��RNA�̊J��ɕ��ՓI�ȃ��[���ƍl����ꂽ�B
�}2a
�}2b
�uAriadne�v(17)�́A�����̌�������b�ɂ��ĕM�҂炪�J���������E�ŏ��߂Ă�RNA�����G���W���ł���i�����o��ρFhttp://ariadne.riken.jp/�j�B���̃\�t�g�E�F�A�́ARNase�Ȃǂɂ���ĉ���z����ٓI�ɐؒf����RNA�f�Ђ��瓾�����A��MS/MS�f�[�^�̃Z�b�g����A����RNA����肷��\�t�g�E�G�A�ł���i�}�R�j�B ���̍ő�̓����́A�������@ MS/MS�f�[�^�ɂ��RNA�f�Ђ̔z�͂Ɠ���iMS/MS�C�I�������j�ƇA ���肵��������RNA�f�Ђ̔z����𗘗p����RNA�̓���i�k�N���I�`�h�}�b�s���O�@�j�̓�i�K�ōs�����Ƃł���B��i�K�ڂ�MS/MS�C�I�������ł́A���ۂ�MS/MS�J��p�^�[�����l�������m�����f���ɂ���Ď���RNA�ɗR������MS/MS�f�[�^���ʓI�ɕ]�����A�ł���v�x�̍���RNA�z��肷��B��i�K�ڂ̃k�N���I�`�h�}�b�s���O�@�ł́AMS/MS�C�I�������œ��肵�������̔z���DB���ł̏o���m���Ɋ�Â��ĕ]�����A����RNA�����肳���m���������_���Ȏ��ۂ����L�ӂɒႢ���ۂ����v�Z���邱�Ƃœ����RNA�肷��B���̃k�N���I�`�h�}�b�s���O�@�̓v���e�I�~�N�X�Ŏg�p����錟���G���W���ɂ͖����Ǝ��̂��̂ł���A���̃A���S���Y�����l�Ă��邱�Ƃł͂��߂č\�������̑��l�������Ȃ�RNA���Q�m�����𗘗p���Ċm���ɓ��肷�邱�Ƃ��ł���悤�ɂȂ����B�܂��A�J��������ǂ��d�˂����݂�Ariadne�ł́A���K�͂�RNA DB�i���J����Ă���ᕪ�qRNA DB�Ȃǁj�����ł͂Ȃ��A�q�g��}�E�X�Ȃǂ̋���ȚM���ރQ�m���ڌ������ē����RNA��ł���悤�ɂȂ��Ă���B���Ȃ݂Ƀq�g�Q�m����ΏۂƂ���Ariadne�̌������Ԃ͌���ł�1�`2 ���Ԓ��x�ł���A���p�I�ɂ��\���Ɏg�p�ł�����̂ƂȂ��Ă���B�܂��A��L�̂Q�i�K�]���@���̗p����Ariadne��RNA���ʔ\�͋ɂ߂č������߁A���݂ł͔|�{�זE�Ȃǂ���v���_�E���@�Ő�������RNP�����̂Ɋ܂܂��RNA�������������̂܂ܒ���LC-MS���͂��A����MS/MS�f�[�^��z��DB�ɑ��Č������邱�ƂŁA�����Ɋ܂܂�镡����RNA�����肷��Ɠ����Ƀ��`�����Ȃǂ̊ȒP�ȓ]�ʌ�C������͂ł���悤�ɂȂ��Ă���B
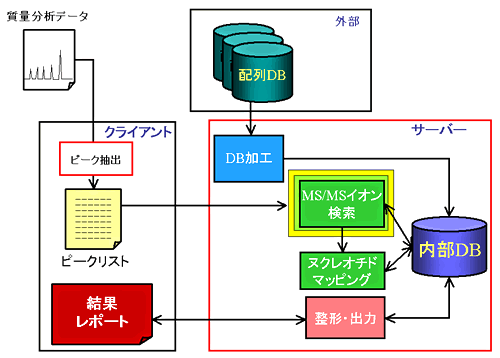
�}3
RNP�����̂̃��{�k�N���I�v���e�I�~�N�X�����FRNA�ƃ^���p�N���̕��s���
�M�҂�́A�זE���ɑ��݂���RNP�����̂��n���I�ɉ�͂��邱�ƂōזE�̋@�\��RNP�����̂̍\��������A�@�\���ߋ@�\�𖾂炩�ɂ���ƂƂ��ɁARNA�ƃ^���p�N���̑��ݍ�p�l�b�g���[�N�Ɋւ���m���邱�Ƃ�ڕW�Ƃ��Ă���B�Ⴆ�A����̃^���p�N�����x�C�g�ɂ��ĖƉu���~�@�ȂǂŐ�������RNP�����̂��\������^���p�N����RNA��������s���Đv���ɓ��肷�邱�ƂŁA�ړI�Ƃ��镡���̂̍\���Ƌ@�\�̃_�C�i�~�N�X�𖾂炩�ɂ��邱�Ƃ��ł��邵�A�@�\���m��RNA�����^���p�N�����܂�RNP�����̂��ł�����̍\�������̓��肪�@�\��͂̒[���ƂȂ�A����ɂ�RNA�|�^���p�N���̑��ݍ�p�Ɋւ���V�����m�������҂ł���B
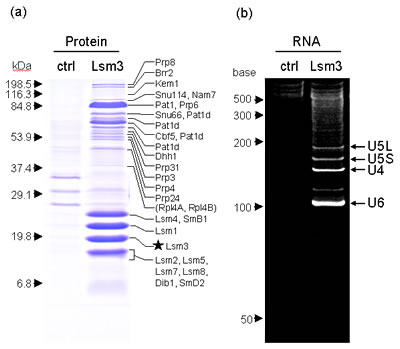
�{�e�ŏЉ��Ariadne��g�ݍ���RNA�̎��ʕ��͖@��K�p���āA�y��̃X�v���C�Z�I�\�[�����̂͂�������}�S���}�T�Ɏ������B���̗�ł́A���i�K�A�t�B�j�e�B�[�^�O�������uLsm-3�^���p�N���v���x�C�g�ɂ��čy��זE�iS.cerevisiae�j���番�����������X�v���C�Z�I�\�[�����̂���t�F�m�[���N�����z�������o�@�ƃA�Z�g�����a�@��g�ݍ��킹�����@�Ń^���p�N����RNA�𒊏o��(13)�A���ꂼ��̐�����d�C�j���@�ŕ��͂����i�}�S�j�B�܂��A�����Ō��o�����^���p�N�������͏]���̃v���e�I�~�N�X�Z�p�œ��肵�ARNA������RNase T1�ɂ��Q�����������LC-MS/MS-Ariadne�V�X�e���œ��肵���i�}�T�j�B�}�Ta �ɂ�RNA�̓d�C�j���Ō��o���ꂽ�����Ƃ������q�ʂ̃o���h�iU4�j��MS/MS���y��Q�m����Ariadne�������ē���ꂽ�uScore histogram�v�������A�}�Tb�ɂ͂��̃o���h��MS/MS�f�[�^���瓯�肵��RNA�f�Ђ��y��Q�m�����\�����邷�ׂĂ̐��F�̏�Ƀ}�b�v�������ʂ��������B������̌��ʂ����̃o���h��U4 snRNA�ł��邱�Ƃm�Ɏ����Ă���B���̌����ł́ALsm3�����̂��\�������30��ނ̊��m�^���p�N����3��ނ̐V�K�^���p�N���ƂƂ��ɁA���̕����̂̍\�������ł���U4�AU5S�AU5L�AU6 �̂S��ނ�snRNA�̂��ׂĂ�����ł����B����ɁA���̕��͂œ���ꂽMS�X�y�N�g�����ڂ�����͂��Ă݂�ƁA������U-snRNA�ɂ́A���m��5�f�L���b�v�\����`�����C���̂ق��ɁA�]���͒m���Ă��Ȃ�3'���[�]�ʏI���ʒu�̕s�ψꐫ�����邱�Ƃ����炩�ɂȂ����B���̕s�ψꐫ�ɂ��ẮA�]���̕��q�����w��זE�����w�̎�@�����p���Ȃ���A���̊m�F����w�I�ȈӋ`�ɂ��Ă̌�����i�߂Ă���B�܂��A�M�҂�͂��̕��@�𗘗p���āA�y���q�g�זE���番�������X�v���C�Z�I�\�[����{�\�[���A�}�C�N��RNA��ӂɊւ�邳�܂��܂�RNP�����̂̉�͂�i�߂Ă���B
�}4
�}5
������
�M�҂�̓v���e�I�~�N�X��RNA�������Z�������u���{�k�N���I�v���e�I�~�N�X�v�������A����̐����Ȋw�̐i�W�ɋɂ߂ďd�v�ł���ƍl���Ă���B�{�e�ŏЉ��RNA�̎��ʕ��͖@�́A�Q�m�����𗘗p����RNA���\���I�ɓ��肵�ē]�ʌ�C�����܂މ��w�\������͂���]���ɂ͂Ȃ��V�����T�O�ƕ��@�_�������̂ł���A�]������s���Ă����t�]�ʔ����𗘗p����RNA��͖@��⊮���郊�{�k�N���I�v���e�I�~�N�X�����̊�b�Z�p�̂P�ƂȂ邱�Ƃ����҂����B�{�e�ŏq�ׂ��悤�ɁA���݂��̕��@�́A�|�{�זE�Ȃǂ��番�������t�F���g�������x����RNP�����̂�RNA�������^���p�N�������Ƃقړ������x�œ��肵�A���w�\������͂ł���悤�ɂȂ��Ă���B����͂���Ȃ鍂���x���ƒ�ʖ@�Ȃǂ��������邱�ƂŁA�זE�@�\�߂��邳�܂��܂�RNP�����̂̉�́A�Ƃ��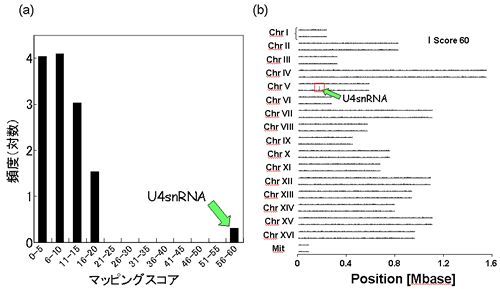 �����݂͂܂��悭�m���Ă��Ȃ��}�C�N��RNA��ӂɊւ��RNP�����̂̐�������@�\���߂Ɋւ��RNA�̏C���╪�q�W���@�\�A����ɂ͍זE�̊��ω��Ɉˑ�����RNP�����̂̃_�C�i�~�N�X�̉�͂�RNA-�^���p�N���̃l�b�g���[�N��͂Ȃǂ̌����Ɋ�^���邱�Ƃ����҂����B
�����݂͂܂��悭�m���Ă��Ȃ��}�C�N��RNA��ӂɊւ��RNP�����̂̐�������@�\���߂Ɋւ��RNA�̏C���╪�q�W���@�\�A����ɂ͍זE�̊��ω��Ɉˑ�����RNP�����̂̃_�C�i�~�N�X�̉�͂�RNA-�^���p�N���̃l�b�g���[�N��͂Ȃǂ̌����Ɋ�^���邱�Ƃ����҂����B
�}�̐���
�}�P�D���{�k�N���I�v���e�I�[���͂̊T�O�}��
�g�D��|�{�זE���琸������RNP�����̂��\������RNA�ƃ^���p�N������s���ē��肵�A�����ɂ��̉��w�\������͂���B
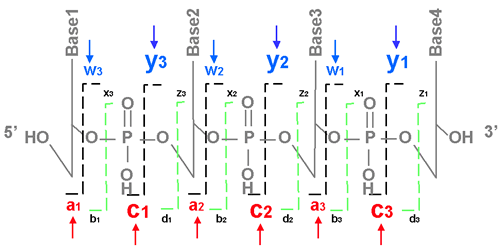
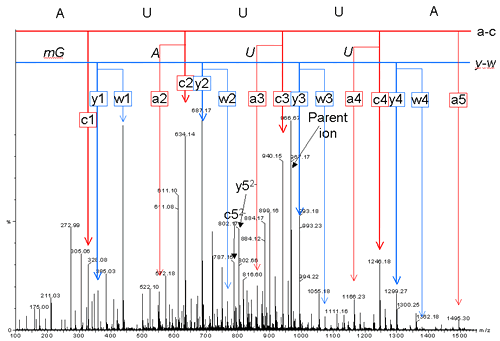
�}�Q�DMS/MS�ɂ�����RNA�̊J��X��
(a) RNA�̊J�����ʂ�͎��I�Ɏ������B�Փˉ𗣌^��MS/MS�ł�c/y�^�C�v�̖��C�I����a/w�^�C�v�̖��C�I����������Bc/y�^�C�v�ɔ��a/w�^�C�v�����キ���o�����X��������Bb/x�Ad/z�^�C�v�̃C�I���͂قƂ�nj��o����Ȃ��B
(b) �y��t�F�j���A���j��tRNA��RNaseT1�����f��5'-AUUUAmG-3'�imG�̓��`���O�A�j���j�̎��ۂ�MS/MS�X�y�N�g���B��ȃV�O�i����c/y�^�C�v��a/w�^�C�v�B
�}�R�D�f�[�^�x�[�X�����G���W��Ariadne�̃V�X�e���\��
�}�S�D�y��X�v���C�Z�I�\�[�����̂̍\����������
Taoka M. et al.: Nucleic Acids Res (2009) doi:10.1093/nar/gkp732�����ς��ē]�ځB(a) Lsm3���x�C�g�Ƃ��ăv���_�E�����������̂̃^���p�N����SDS-PAGE�B�^���f���A�t�B�j�e�B�[�^�O���g����2�i�K�����ɂ���ē���ꂽ�^���p�N�����j����N�}�V�[���F�����B��ȃ^���p�N�������̓��茋�ʂ̓��[���E���Ɏ������B(b) ���������̂Ɋ܂܂��RNA��Urea-PAGE�B���F��SYBR Gold�ɂ��BRNA�̓��茋�ʂ̓��[���E���ɒ����B���̑��A�ڂ����͕������Q�ƁB
�}�T�DLC-MS-Ariadne�ɂ��X�v���C�Z�I�\�[�������̂Ɋ܂܂��U4 snRNA�̓���
�}�Sb�Ɏ������uU4�v�̃o���h�����ALC-MS�ŕ��͂���Ariadne�ʼn�͂��AU4snRNA�肵���B(a)�͓���̍ۂ̃k�N���I�`�h�}�b�s���O�̃X�R�A�̕p�x���z�A(b)�̓o���hU4��MS/MS�f�[�^���瓯�肵��RNA�f�Ђ��y����F�̏�Ƀ}�b�v�������ʁB�y��Q�m����Ƀ}�b�v���ꂽRNA�f�ЌQ�̈ʒu��U4snRNA���R�[�h����Ă���ʒu����v���Ă���B
�L�[���[�h
�ELC-MS �ERNA �E���{�k�N���I�v���e�I�~�N�X
�ӎ�
�{������JST ���x������ CREST �����̎����ɂ����̂ł���B���̌��e�������ɓ�����A�����͂����������R���F�Y���m�A���D�q���A���؏r�����m�Ɋ��ӂ������܂��B
����
- Juhling F. et al.: Nucleic Acids Res (2009) 37: D159-162.
- Goto-Ito S. et al.: Nat Struct Mol Biol (2009) 16: 1109-1115.
- Kaneko T. et al.: EMBO J (2003) 22: 657-667.
- Okamoto S. et al.: Mol Microbiol (2007) 63: 1096-1106.
- Banoub J. H. et al.: Chem Rev (2005) 105: 1869-1915.
- Noma A. et al.: �`�����j�_�y�f (2006) 51: 2226-2231.
- Noma A. et al.: EMBO J (2006) 25: 2142-2154.
- Suzuki T. et al.: EMBO J (2002) 21: 6581-6589.
- Ohara T. et al.: Nat Struct Mol Biol (2007) 14: 349-350.
- Miyauchi K. et al.: Nucleic Acids Res (2007) 35: e24.
- Katoh T. et al.: Genes Dev (2009) 23: 433-438.
- Natsume T. et al.: Anal Chem (2002) 74: 4725-4733.
- Taoka M. et al.: Nucleic Acids Res (2009) doi:10.1093/nar/gkp732.
- Huang T. Y. et al.: J Am Soc Mass Spectrom (2008) 19: 1832-1840.
- Schurch S. et al.: J Am Soc Mass Spectrom (2002) 13: 936-945.
- Douthwaite S. et al.: Methods Enzymol (2007) 425: 1-20.
- Nakayama H. et al.: Nucleic Acids Res (2009) 37: e47.